First-principles calculations of alloy phase diagrams using Machine Learning
The Machine-learning Interatomic Potentials (MLIPs) can be used to significantly accelerate the process of phase diagram construction for alloys by providing a surrogate model interpolating the quantum-mechanical data. To construct an alloy phase diagram a model calculating the properties of atomic systems is required. Typical way is to use ab-initio methods such as DFT to calculate energies, forces and stresses. However, ab-initio calculations require a lot of CPU time and thus the whole process can take a long time to be completed. At the same time, while running a molecular dynamics (MD) simulation or a structure relaxation a lot of similar atomic configuration occur. In our approach we use machine learning (ML) techniques to infer the hidden relations between geometric structure and composition of an atomic system and its properties, which allows to calculate the properties of the majority of occurring atomic systems with MLIPs, which is orders of magnitude faster than with DFT, without significant loss of accuracy.
Building the convex hull without ML would take at least 5000 times more CPU time
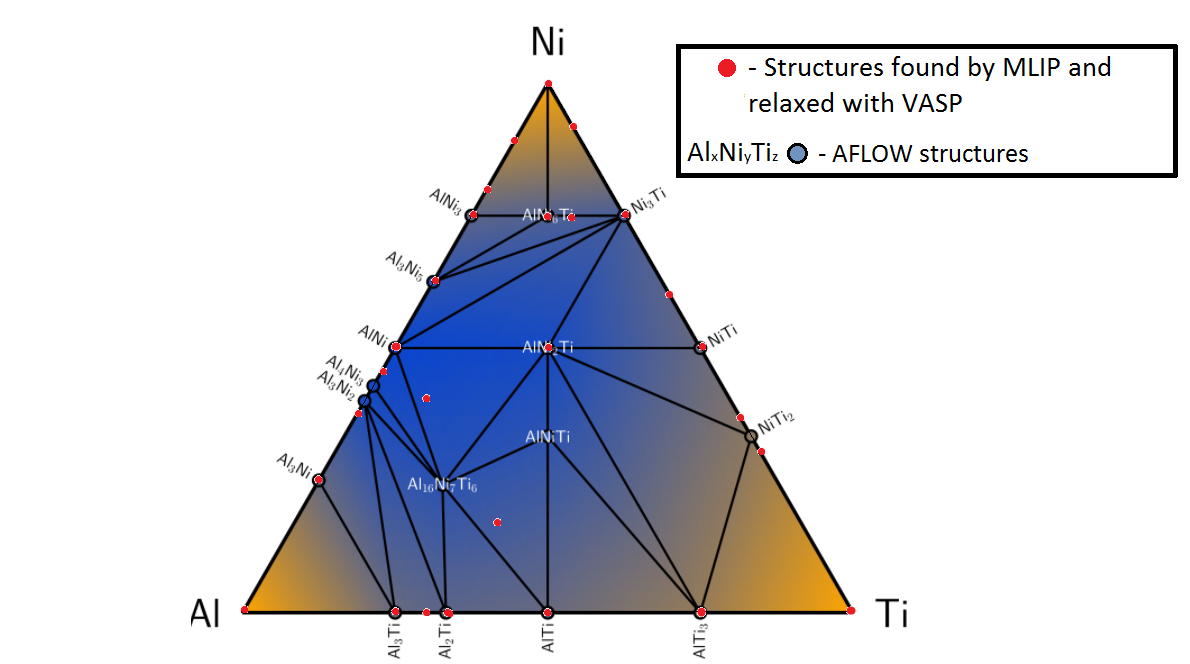
Al-Ni-Ti convex hull
One of the proof-of-concept applications of MLIP is predicting the stable structures of metallic alloys. To verify our approach we have built the convex hulls for Cu-Pd, Co-Nb-V and Al-Ni-Ti systems. In each case we selected some amount of candidate crystal structures and performed relaxation of their structure with the further calculation of formation energies for relaxed systems. Employing a MLIP allows to pick much greater pool of initial samples for relaxation in comparison to when DFT only is used. Most of these structures are relaxed with a MLIP, while the implemented active learning algorithm picks relatively small amount of structures to be calculated with DFT and form a training set.
In the case of Al-Ni-Ti alloy system, after training on several hundreds of configurations with ab-initio energies, forces and stresses MTP was able to relax hundreds of thousands of candidate structures. On the picture below you can see the comparison of the convex hulls for Al-Ni-Ti system obtained with MTP and with DFT (this was taken from the AFLOW library). Apart from the finite accuracy of MTP calculations, the mismatch between the convex hulls is also caused by absence of some specific candidate structures from initial MTP pool. For instance, in this test we took candidate structures with not more than 12 atoms in the unit cell, while in the AFLOW convex hull there are some structures containing 56 and 29 atoms in the unit cell.