Machine Learning for Computational Materials Discovery
Crystal Structure Prediction
Discovery of novel solid materials is related to the problem of finding stable atomic structures of given chemical content. As a rule, such stable structures have lowest energy among all possible structures. Unfortunately, the complexity of the problem of crystal structure prediction grows exponentially with the number of atom types and the total number of atoms in the supercell. The most efficient method for solving this problem is USPEX, a code developed by Artem Oganov’s research group. This method is based on a genetic optimization algorithm of finding structures with the lowest energy. For calculation of energy it uses the density functional theory (DFT). DFT calculation takes, typically, more than 99% of the USPEX’s CPU time. Thus, DFT calculations are the bottleneck of the USPEX algorithm, making it impractical for structures with a large number of atoms in the supercell (more than 50).
Application of machine learning models of interatomic interaction
Replacing DFT by computationally efficient Machine Learning Interatomic Potentials that we develop accelerate the crystal structure prediction by orders of magnitude. This allows us to find structures with a large number of atoms in the supercell, which was not possible with DFT. For example, we can predict many-atom Boron structures:


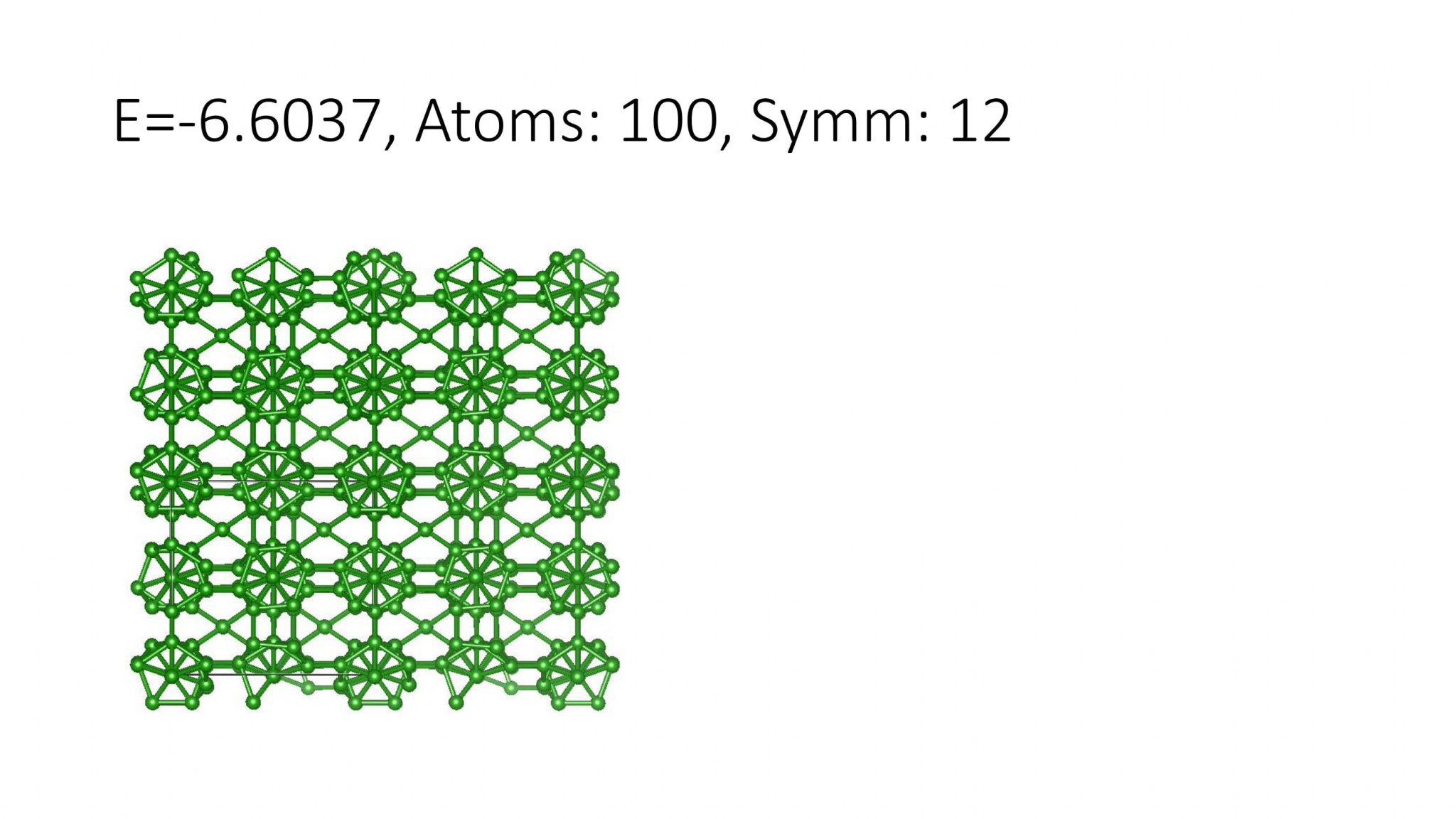
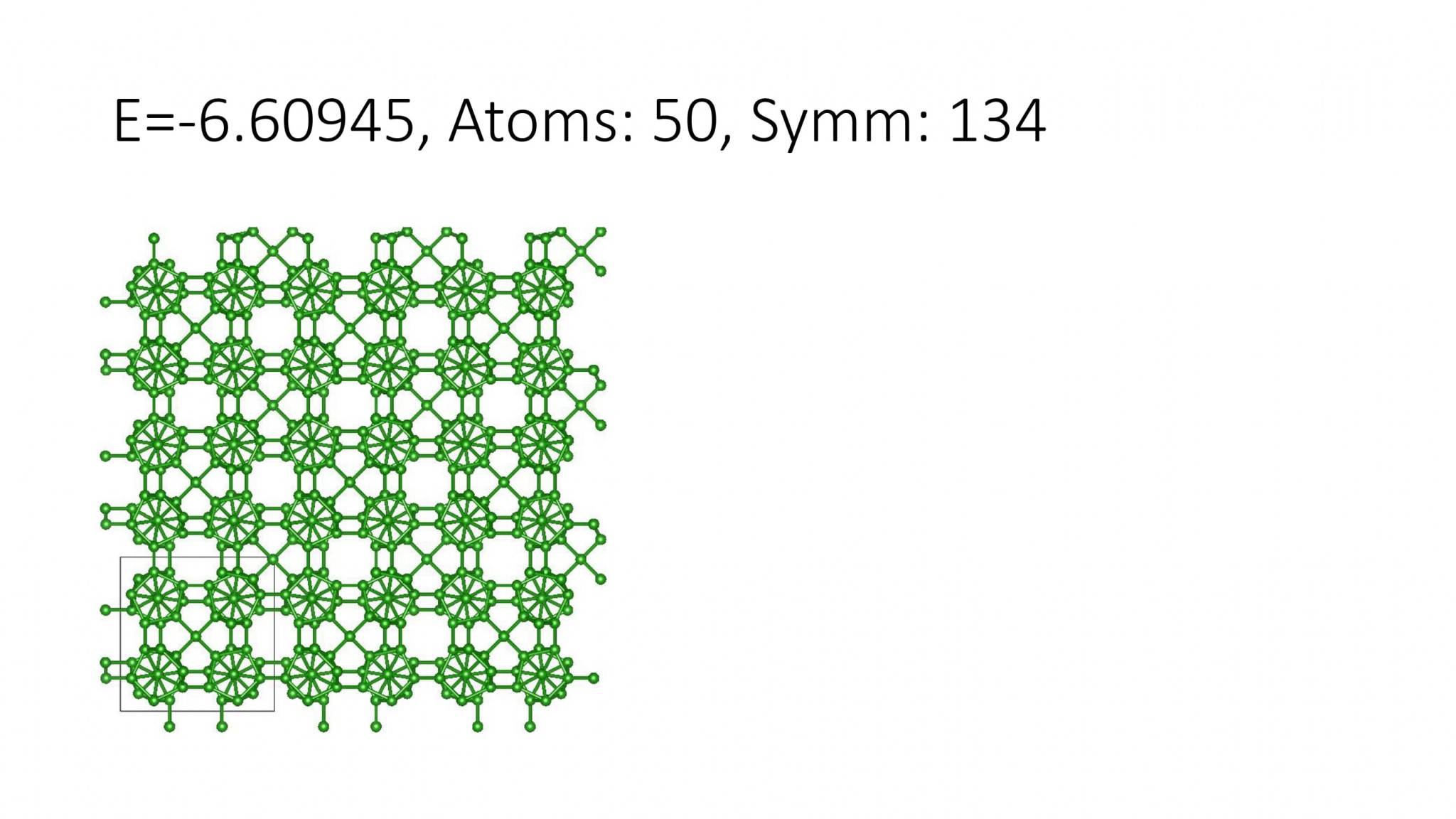
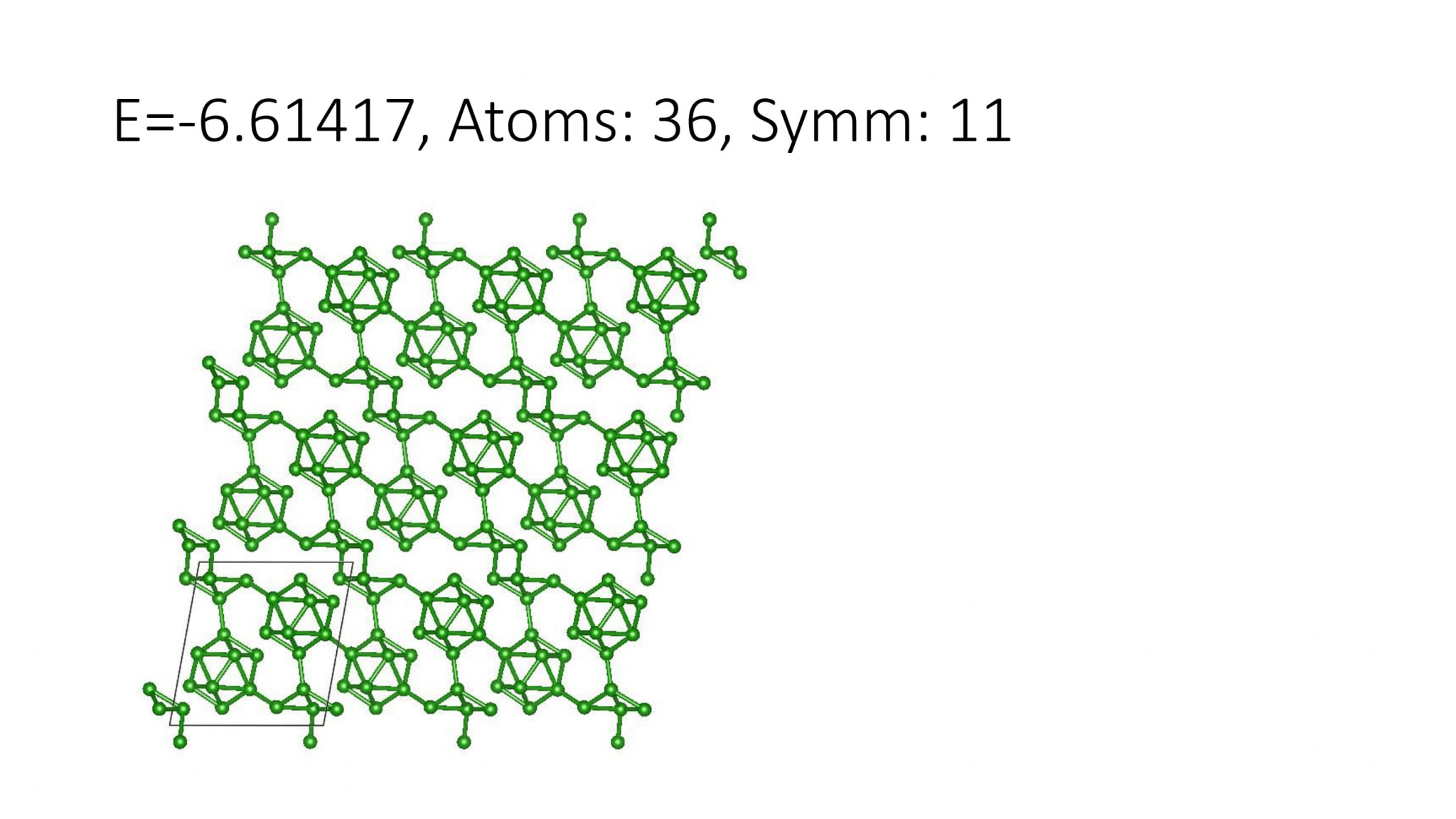
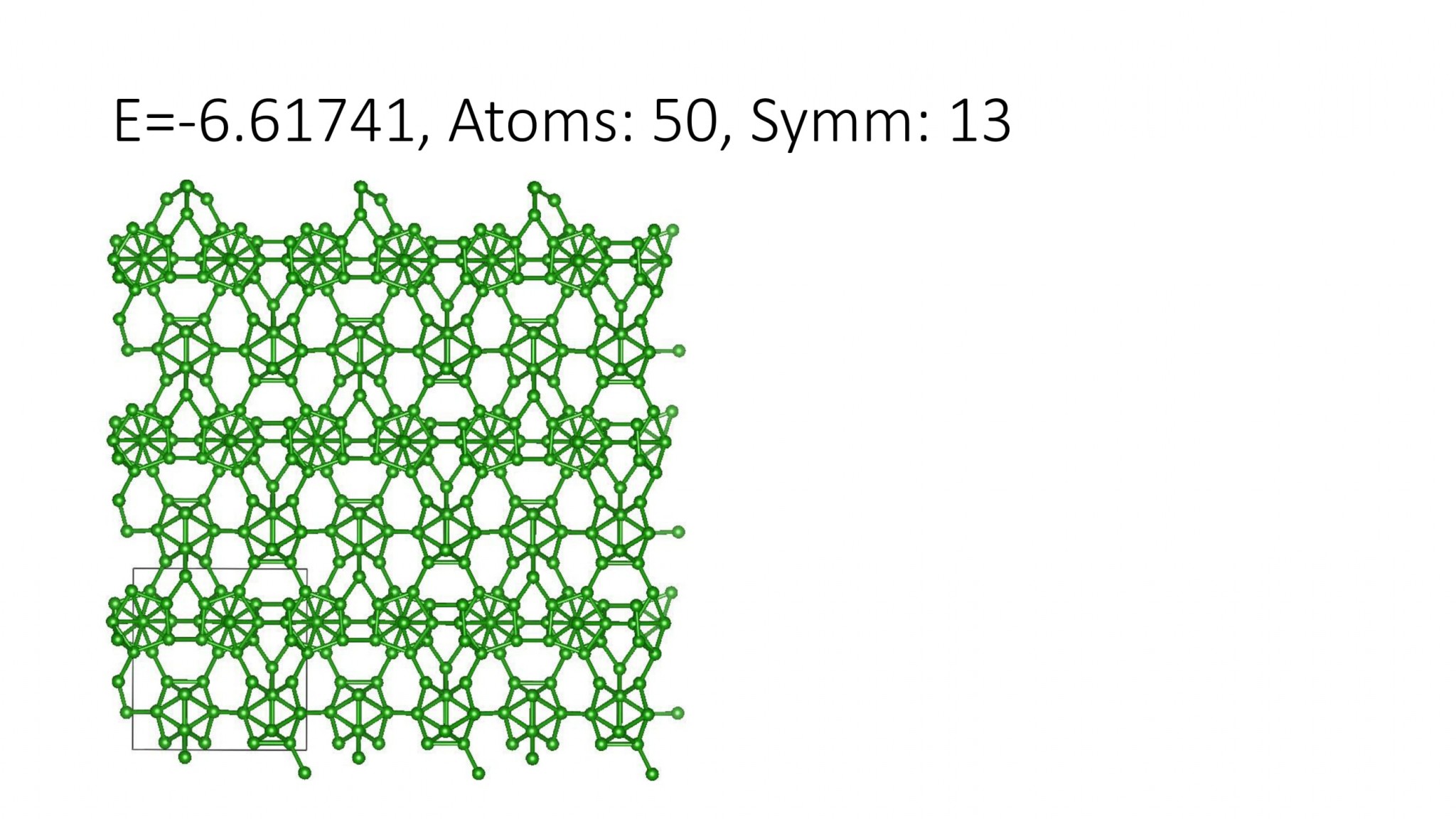
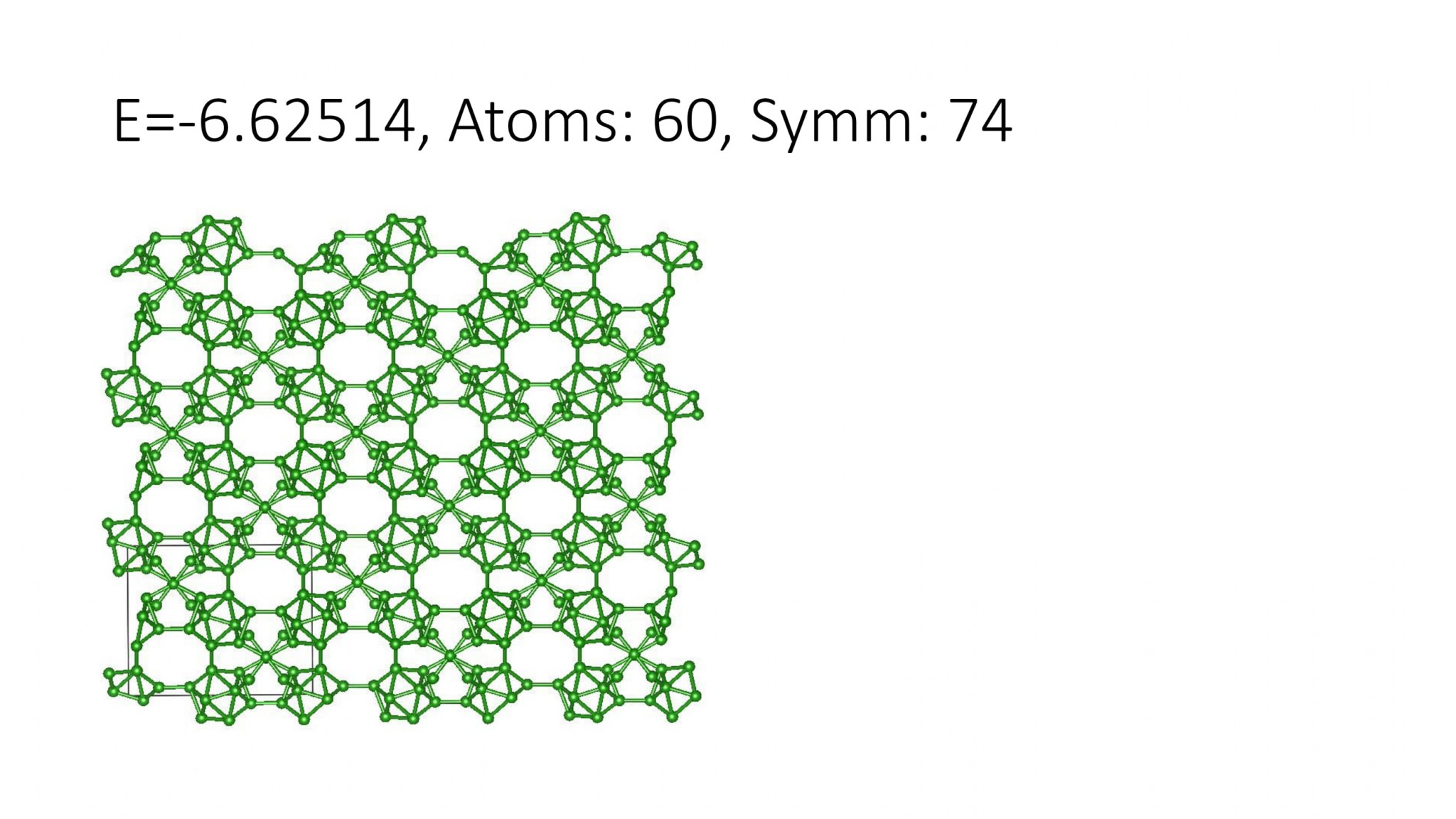



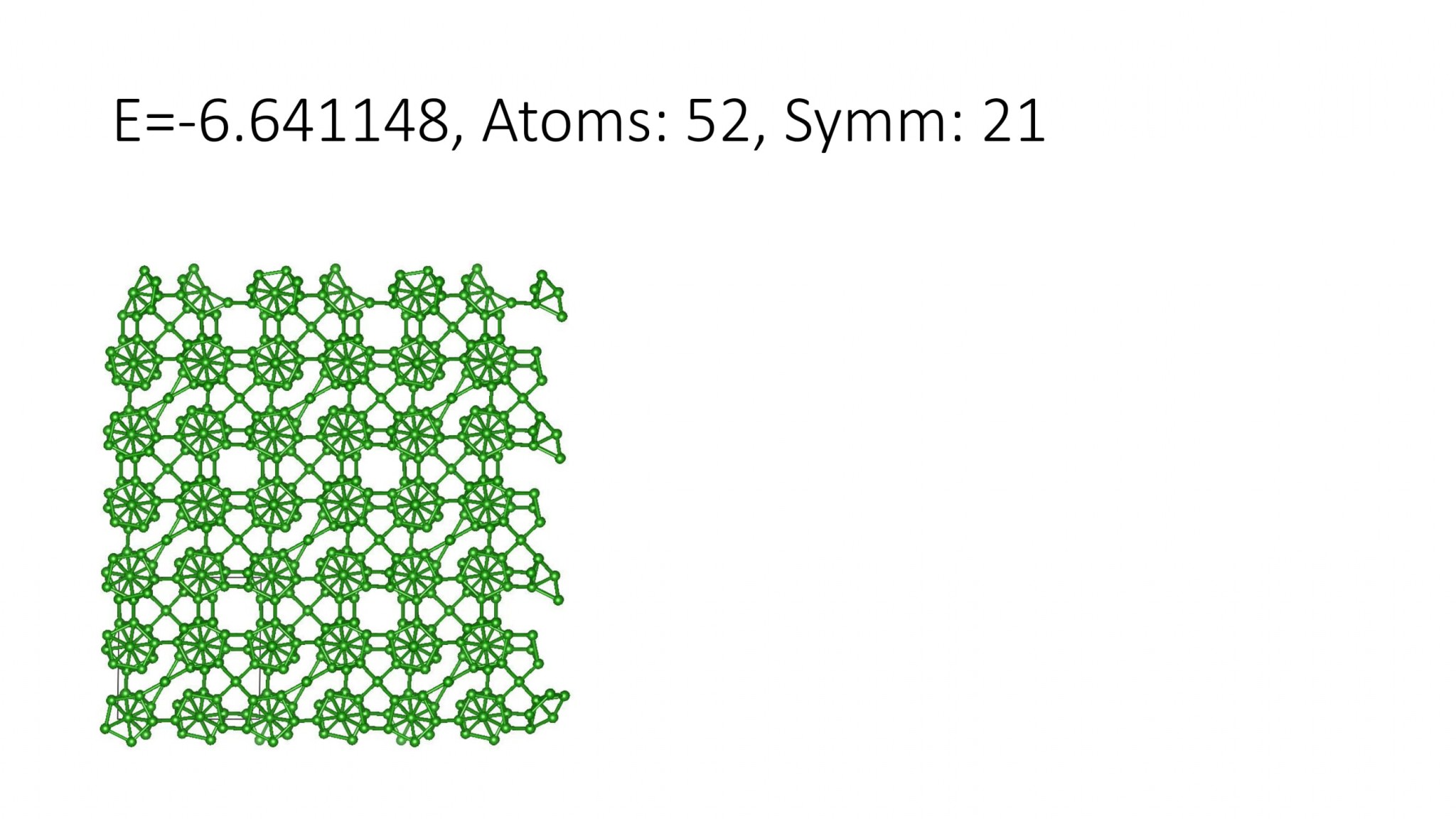

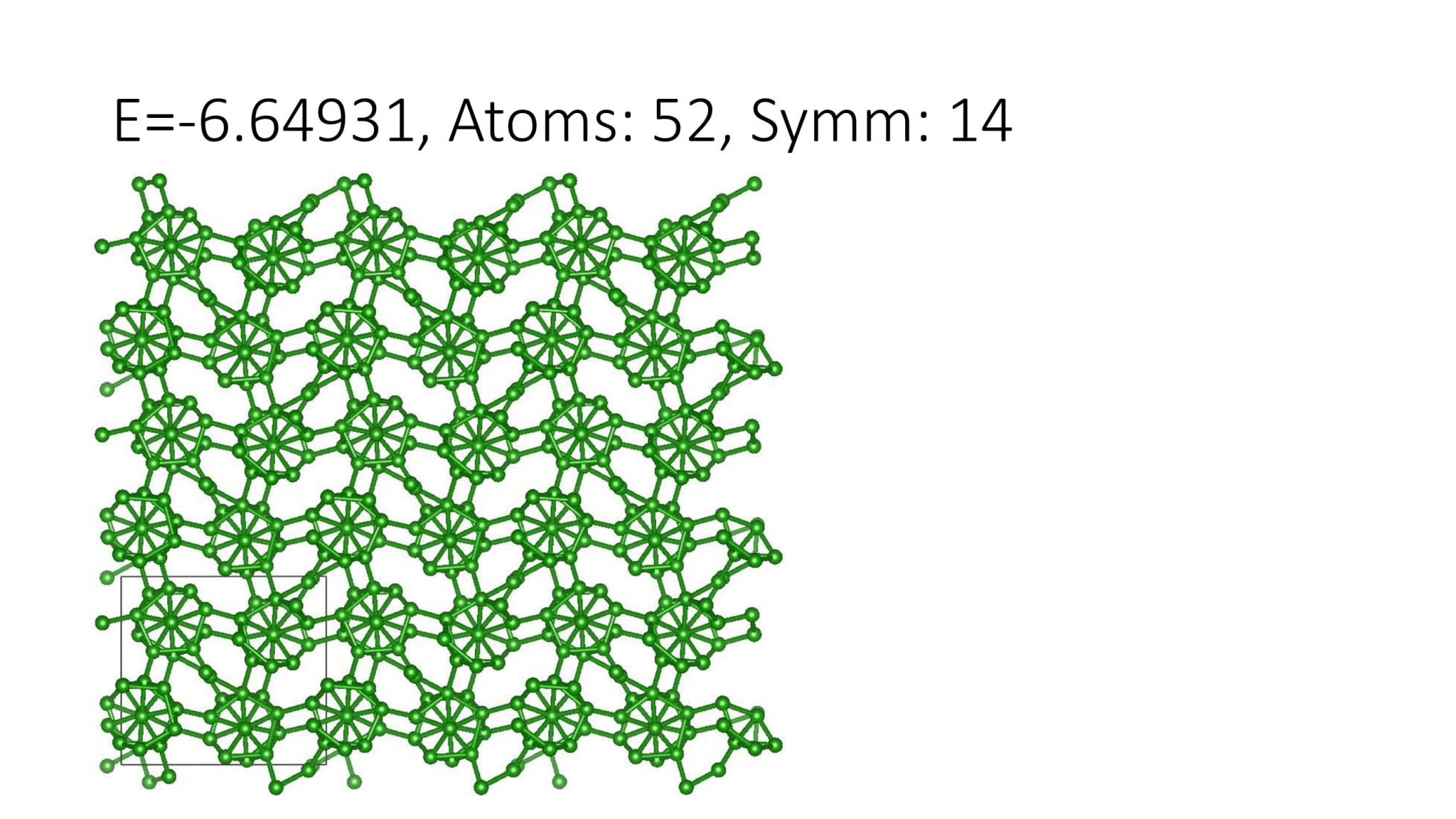
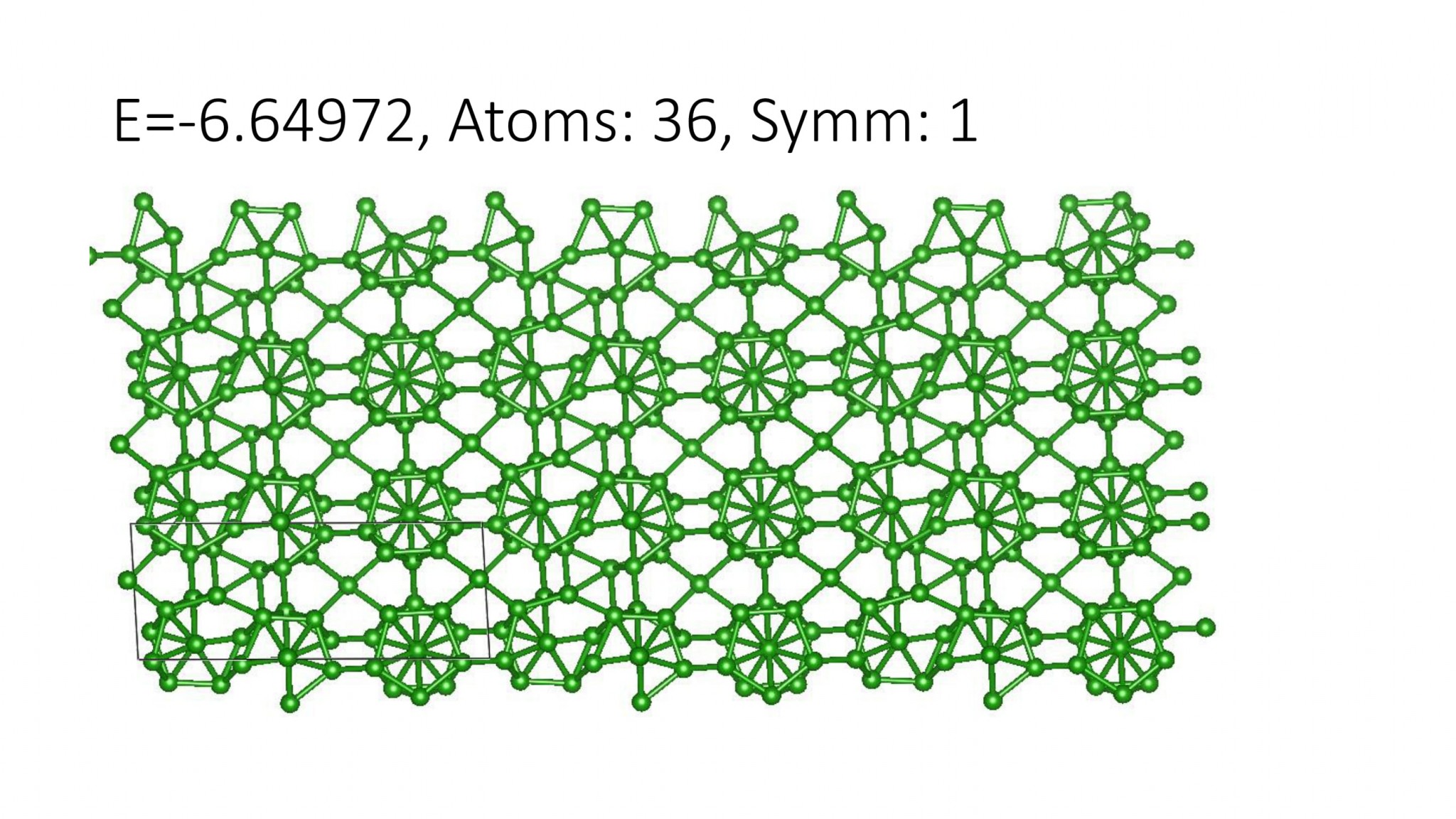
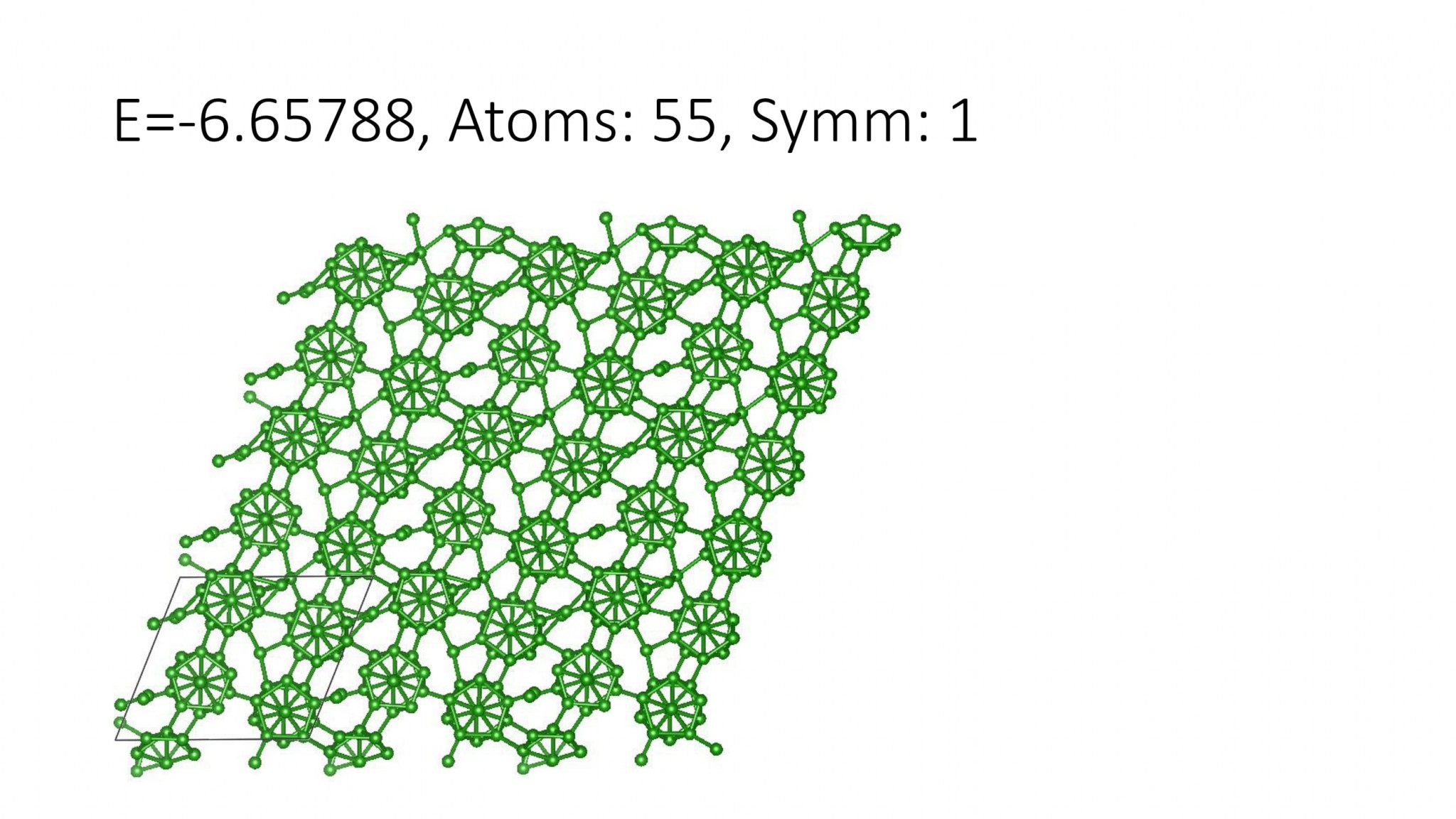
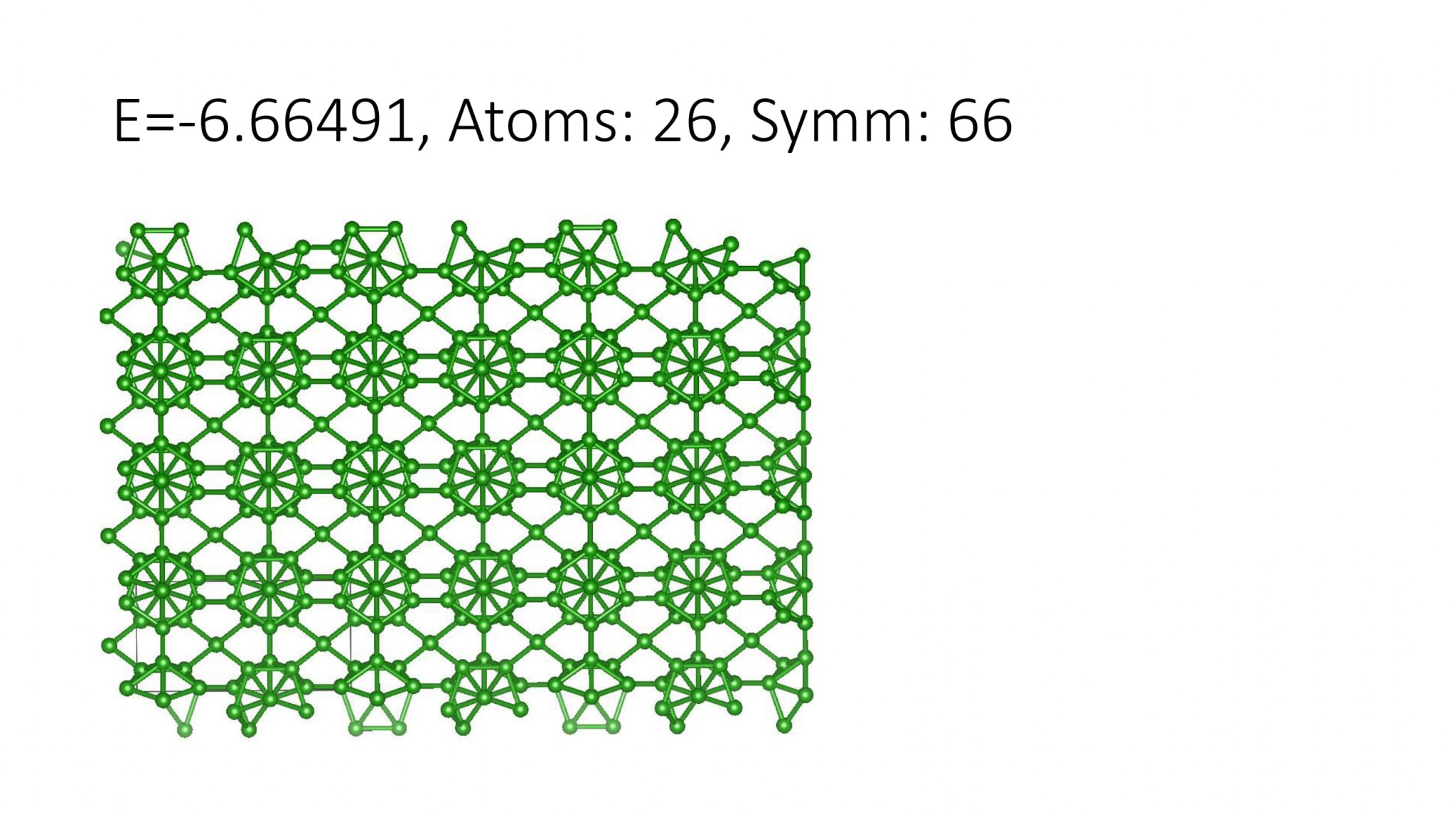



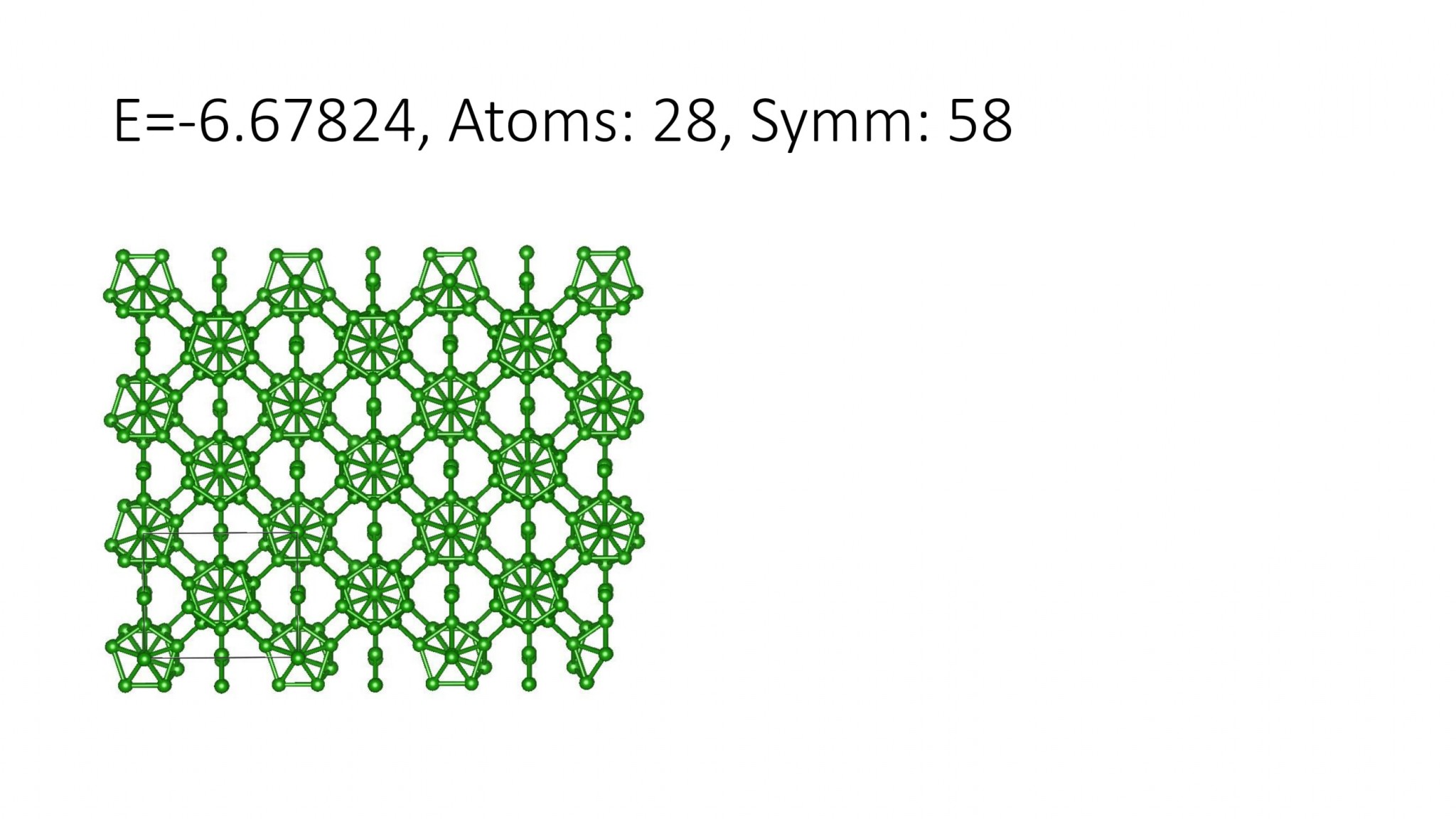
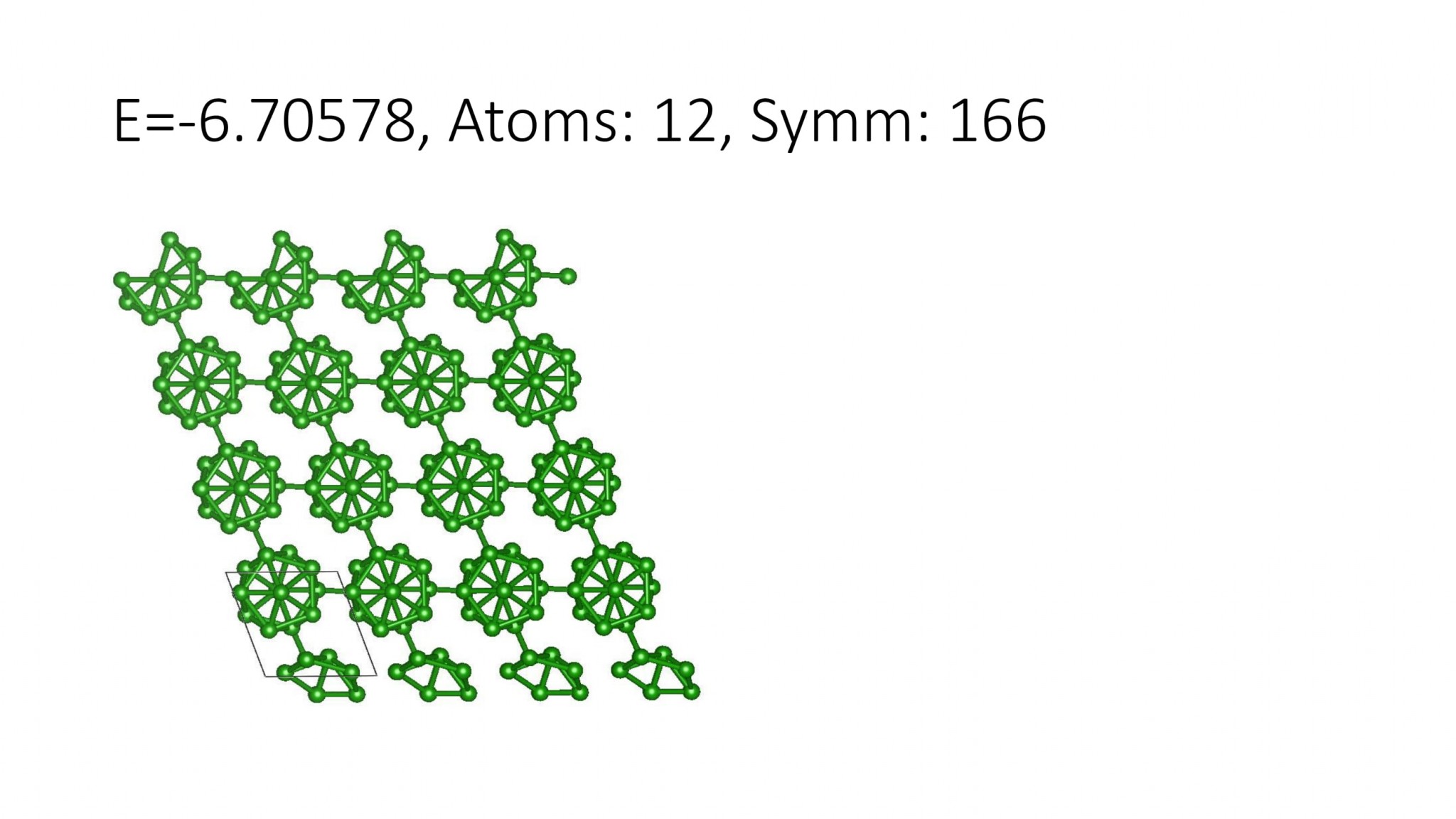
Boron structures found with a Machine Learning model of interatomic interaction