ML potentials in application to polycrystalline and composite materials
Mechanical Properties of Single and Polycrystalline Solids from Machine Learning (Adv. Theor. Sim., 2024, 2301171) PDF
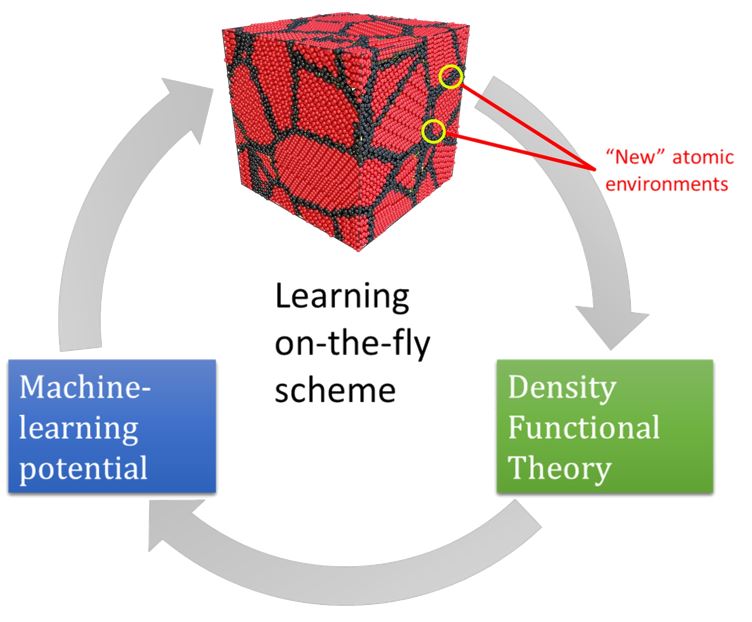
Calculating the elastic and mechanical characteristics of non-crystalline solids can be challenging due to the high computational cost of ab initio methods and the low accuracy of empirical potentials. This paper proposes a computational technique for efficient calculations of mechanical properties of polycrystals, composites, and multi-phase systems from atomistic simulations with high accuracy and reasonable computational cost. The calculated elastic moduli of polycrystalline diamond and their dependence on grain size are determined using a developed approach based on actively learned machine learning interatomic potentials (MLIPs). These potentials are trained on local fragments of the polycrystalline system, and ab initio calculations are used to compute forces, stresses, and energies. This technique allows researchers to perform extensive calculations of the mechanical properties of complex solids with different compositions and structures, achieving high accuracy and facilitating the transition from ideal (single crystal) systems to more realistic ones.
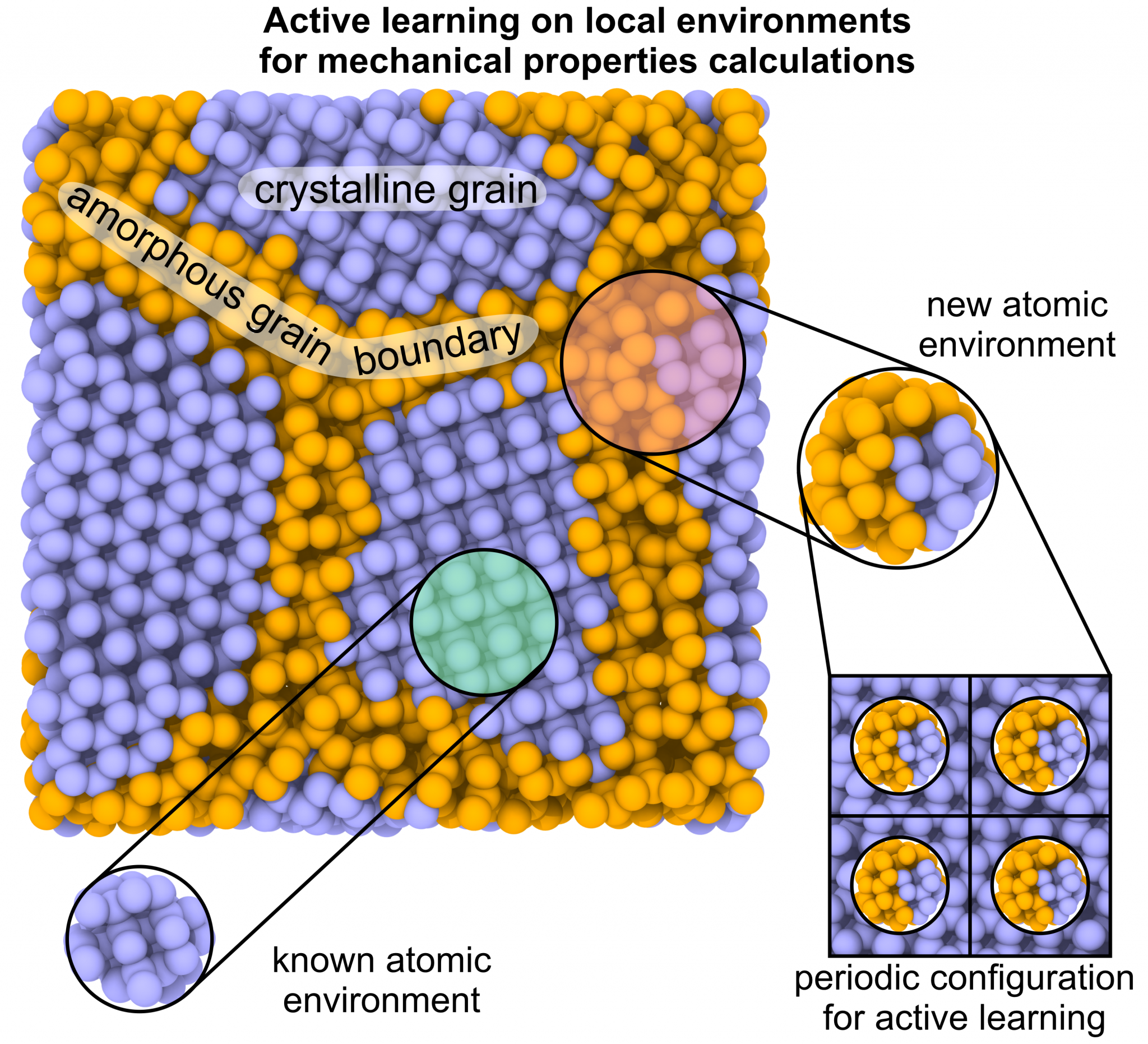
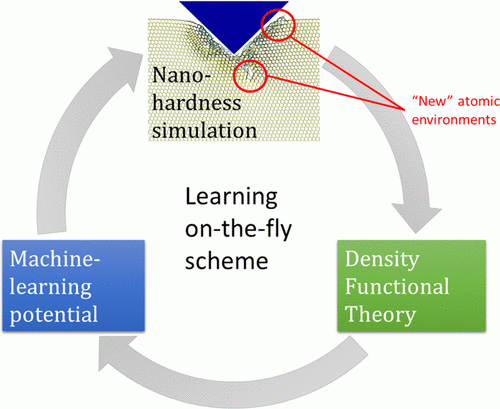
Nanohardness from First Principles with Active Learning on Atomic Environments (J. Chem. Theory Comput. 2022, 18, 2, 1109–1121) PDF
We propose a methodology for the calculation of nanohardness by atomistic simulations of nanoindentation. The methodology is enabled by machine-learning interatomic potentials fitted on the fly to quantum-mechanical calculations of local fragments of the large nanoindentation simulation. We test our methodology by calculating nanohardness, as a function of load and crystallographic orientation of the surface, of diamond, AlN, SiC, BC2N, and Si and comparing it to the calibrated values of the macro- and microhardness. The observed agreement between the computational and experimental results from the literature provides evidence that our method has sufficient predictive power to open up the possibility of designing materials with exceptional hardness directly from first principles. It will be especially valuable at the nanoscale where the experimental measurements are difficult, while empirical models fitted to macrohardness are, as a rule, inapplicable.
{kind=link}