Methods for structure investigations
Physically intuitive anisotropic model of hardness (Phys. Rev. Materials, 2024, 8, 123601)
The hardness of materials plays an important role in material design. There are numerous experimental methods for measuring the hardness of materials, but theoretical prediction of hardness is challenging. By studying the correlation between hardness and the elastic properties of materials, namely shear and bulk moduli, the pressure derivative of the bulk modulus, we have constructed a simple and physically intuitive hardness model. By introducing the spatial variation of the shear modulus, it is possible to predict the hardness anisotropy of materials to define the minimum and maximum values of hardness possessed by a given material. Furthermore, by using the equation of states to define the pressure derivative of the bulk modulus, it is possible to determine the temperature dependence of hardness for given materials. All quantities in the model can be obtained directly from accurate first-principles calculations or from experiments, making it suitable for practical applications.

Chemical bonding within AIIIBVI materials under uniaxial compression (Phys. Chem. Chem. Phys., 2024, 26, 20984-20992)
The work provides a comprehensive explanation of the nature of chemical bonding through quantum chemical topology for multilayers of AIIIBVI compounds, such as GaSe, InSe, and GaTe, spanning pressures from 0 GPa to 30 GPa. These compounds are subjected to pressure orthogonal to the multilayers. Quantum chemical topological indices indicate that uniaxial pressure induces changes in hybridisation, leading to the disappearance of interlayer van der Waals forces. The distinct nature of the elements within the compounds results in different pressures at which van der Waals interactions disappear, as revealed by non-covalent interaction analysis. The presence or absence of chemical bonding is assessed by quantum topological indices as Espinosa indices, charge density distribution difference, and crystal orbital Hamilton populations. The varying changes in hybridisation, as indicated by topological indices, are corroborated by variations in the population of the electronic projected density of states. Ultimately, the type of chemical bonding is identified through the Espinosa indices in the field of Bader theory. This analysis confirms the existence of shared shell bonds between AIII and BVI atoms in vacuum that goes to an intermediate bond between shared and closed shells called the transition zone with increasing pressure. The implications and importance of this work extend beyond the presented results. It suggests that many other classes of two-dimensional materials may undergo phase transitions under uniaxial stress, leading to the formation of new phases with potentially interesting electronic properties.

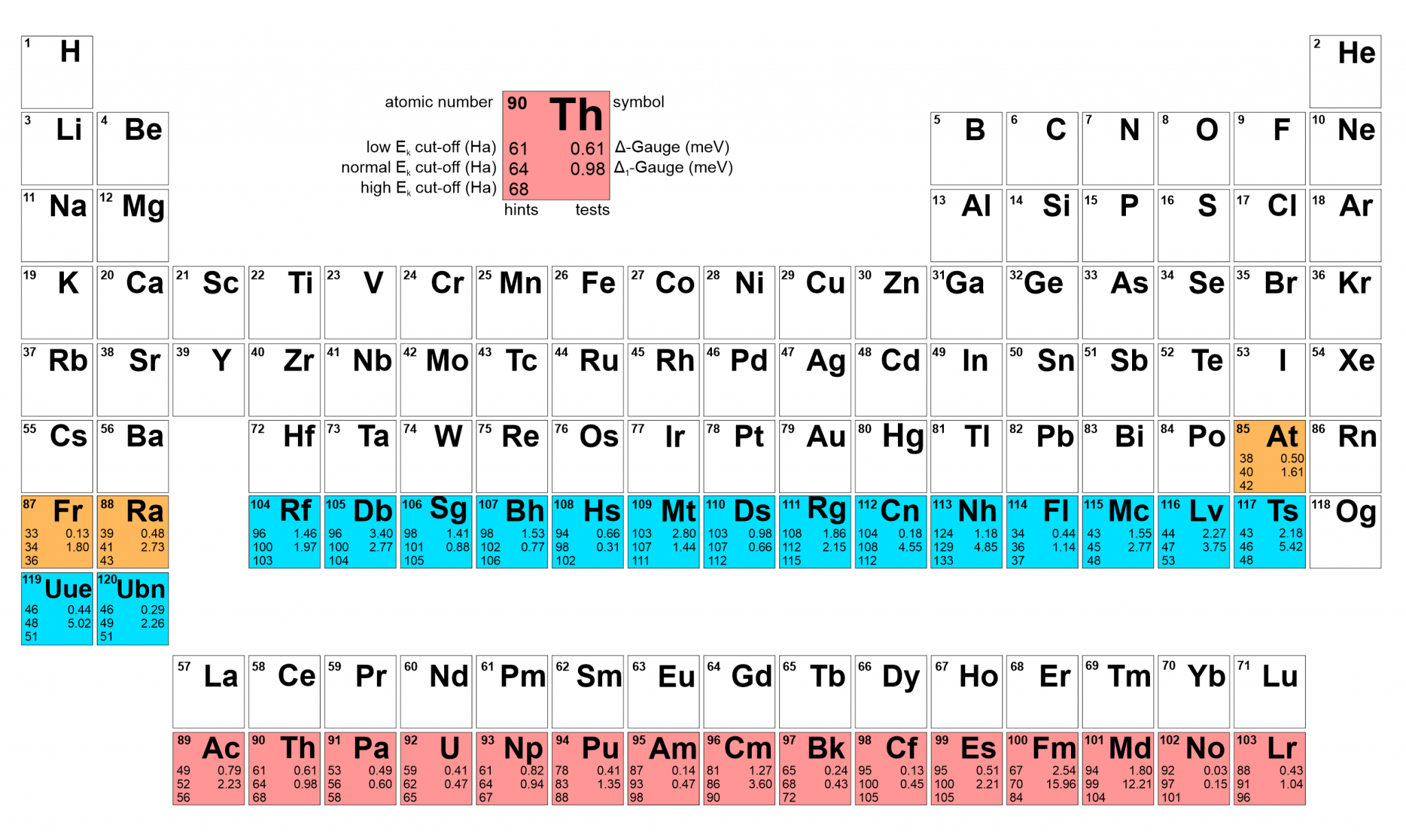
ONCV pseudopotentials for super heavy elements (Comp. Phys. Comm. 2023, 295, 109002)
In this work, we present a series of fully-relativistic optimised norm-conserving Vanderbilt pseudopotentials (ONCVPs) for thirty-four actinides and super-heavy elements. The scalar relativistic version of these ONCVPs is tested by comparing equations of states for crystals, obtained with abinit 9.6, with those obtained by all-electron zeroth-order regular approximation (ZORA) calculations performed with the Amsterdam Modelling Suite BAND code. Δ-Gauge and Δ1-Gauge indicators are used to validate these pseudopotentials. This work is a contribution to the PseudoDojo project, in which pseudopotentials for the whole periodic table are developed and systematically tested. The fully-relativistic pseudopotential files (i.e. including spin-orbit coupling) are available on the PseudoDojo web-interface this http URL under the name NC FR (ONCVPSP) v4.x. Pseudopotentials are made available in psp8 and UPF2 formats, both convenient for abinit, the latter being also suitable for Quantum ESPRESSO.
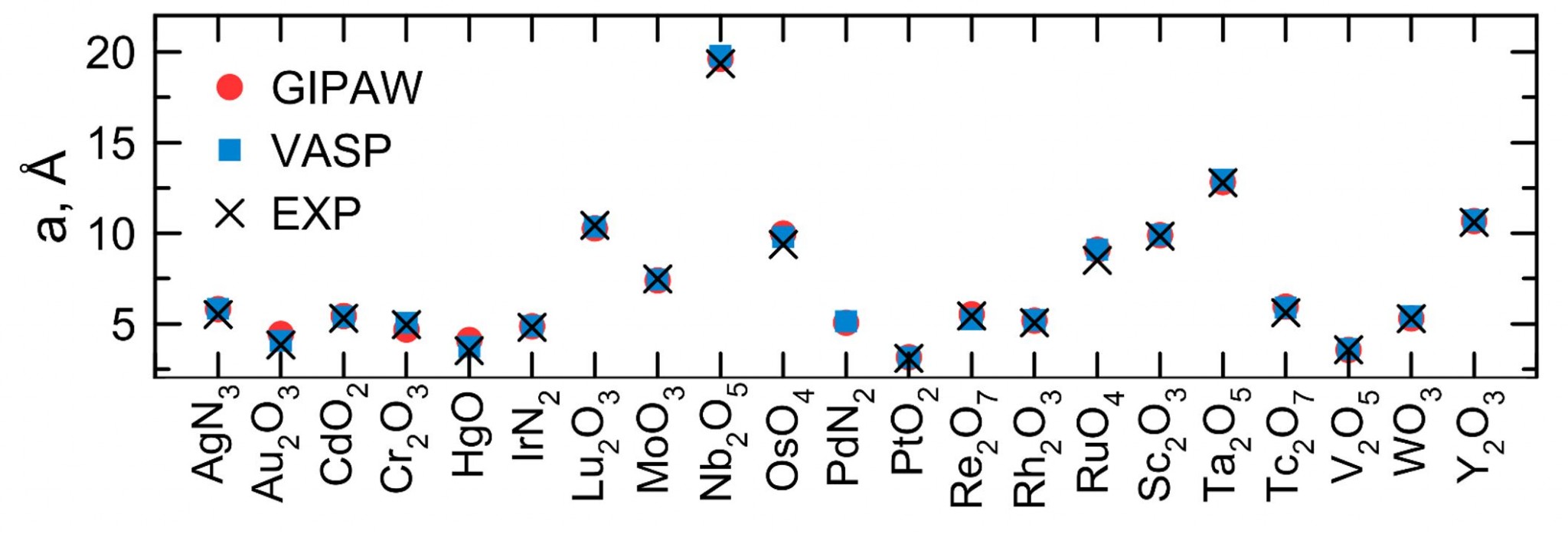
GIPAW pseudopotentials (Materials 2022, 15 (9), 3347)
We developed a gauge including projected augmented pseudopotentials (GIPAW) of 21 d elements and tested them on, respectively, oxides or nitrides (semiconductors), calculating chemical shift and quadrupolar coupling constant. This work can be considered the first step to improving the ab initio prediction of nuclear magnetic resonance (NMR) parameters, and leaves open the possibility for inorganic compounds to constitute an alternative standard compound, with respect to tetramethylsilane, to calculate the chemical shift. Furthermore, this work represents the possibility to obtain results from first-principles calculations, to train a machine-learning model to solve or refine structures using predicted nuclear magnetic resonance spectra.
Link to download developed GIPAWs
More GIPAWs you can find here
Accurate description of strongly correlated fluorine-based compounds (J. Phys. Chem. C, 2022)
We have found an excellent solution to investigate fluorinated materials using a combination of SCAN (exchange) and rVV10 (correlation) functionals. This was found through the fundamental study of α- and β-fluorine phases showing α-fluorine as the most stable structure at the temperatures lower than 35 K and 0 GPa respect to β-fluorine. Further, we have computed crystal structure evolution under pressure looking for new stable fluorine allotropes using USPEX evolutionary algorithm coupled with SCAN-rVV10 exchange-correlation functional discovering two phase transitions.

{kind=link}